Research
Reseach Area 1. First Principles & Computational Materials Science
1. Quasiparticle phonons analysis with neural network potential
Current research at Columbia University as a master’s student - to be added
Keywords: phonons, deep potential, LAMMPS, Snakemake, HPC
2. Monte Carlo geometry optimization and first-principles nanoparticles study
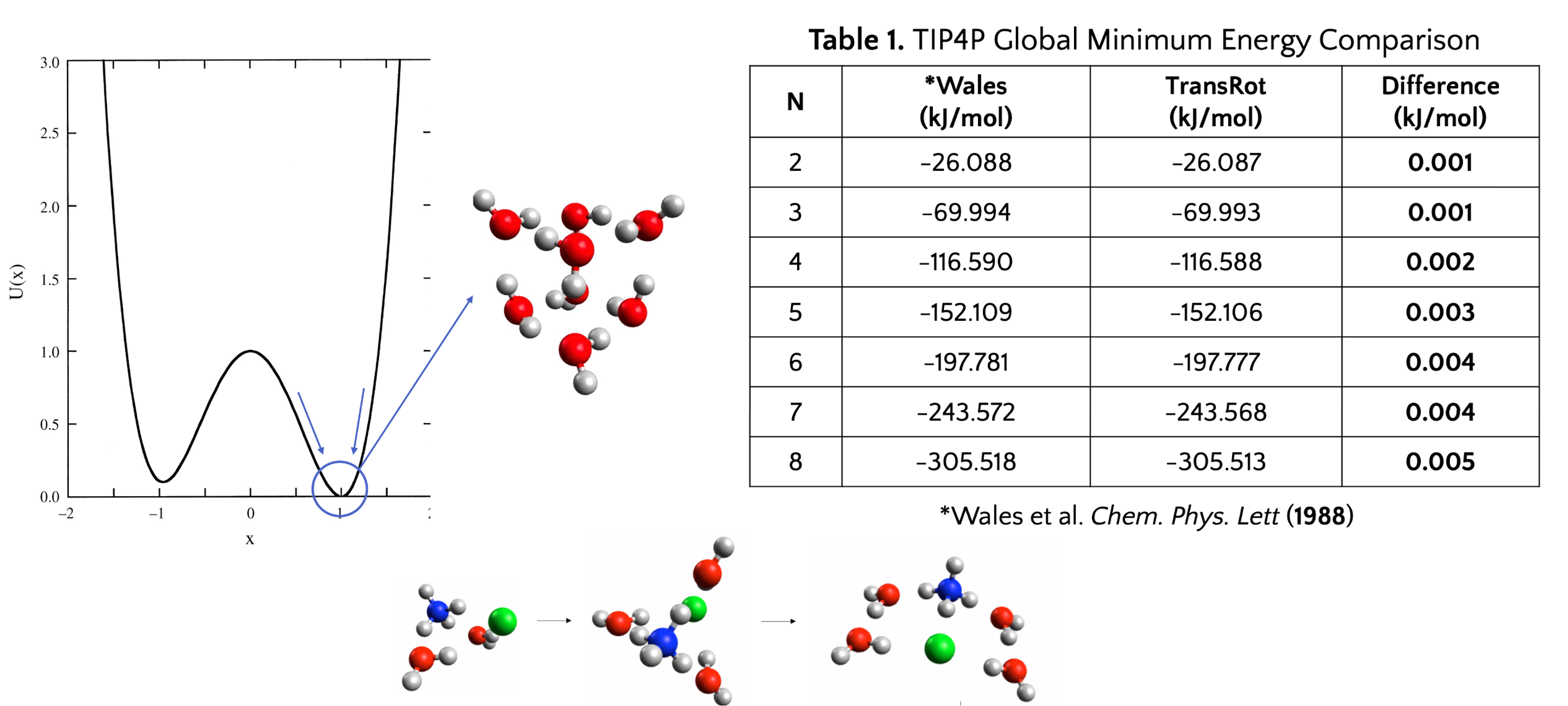 From January 2022 to May 2023, I
conducted three semesters of independent computational chemistry research with
Dr. Robert Topper at the Chemistry Department at The Cooper Union. I was
involved in the group’s focus on using theoretical and computational tools to
study the formation behavior of salt nanoparticles. I focused on the formation
behavior of ammonium chloride solvated in two to eight water molecules.
From January 2022 to May 2023, I
conducted three semesters of independent computational chemistry research with
Dr. Robert Topper at the Chemistry Department at The Cooper Union. I was
involved in the group’s focus on using theoretical and computational tools to
study the formation behavior of salt nanoparticles. I focused on the formation
behavior of ammonium chloride solvated in two to eight water molecules.
For geometry optimization, the group developed an open-source geometry optimizer called “TransRot”. I validated the accuracy and reliability of the open-source software by implementing water models such as TIP4P and TIP4P/2005. I used the TransRot software and conducted ab initio calculations in Psi4, ORCA, and SPARTAN to study the formation behavior with ammonium by determining the binding energy as one water molecule is added to the complex.
Book chapter | Poster | GitHub
Keywords: optimizer, Monte Carlo, ab initio, TIP4P, Psi4, ORCA
Research Area 2. Data-driven Methods for Materials Discovery
1. High-throughput geometric crystal featurizer for binary and ternary compounds
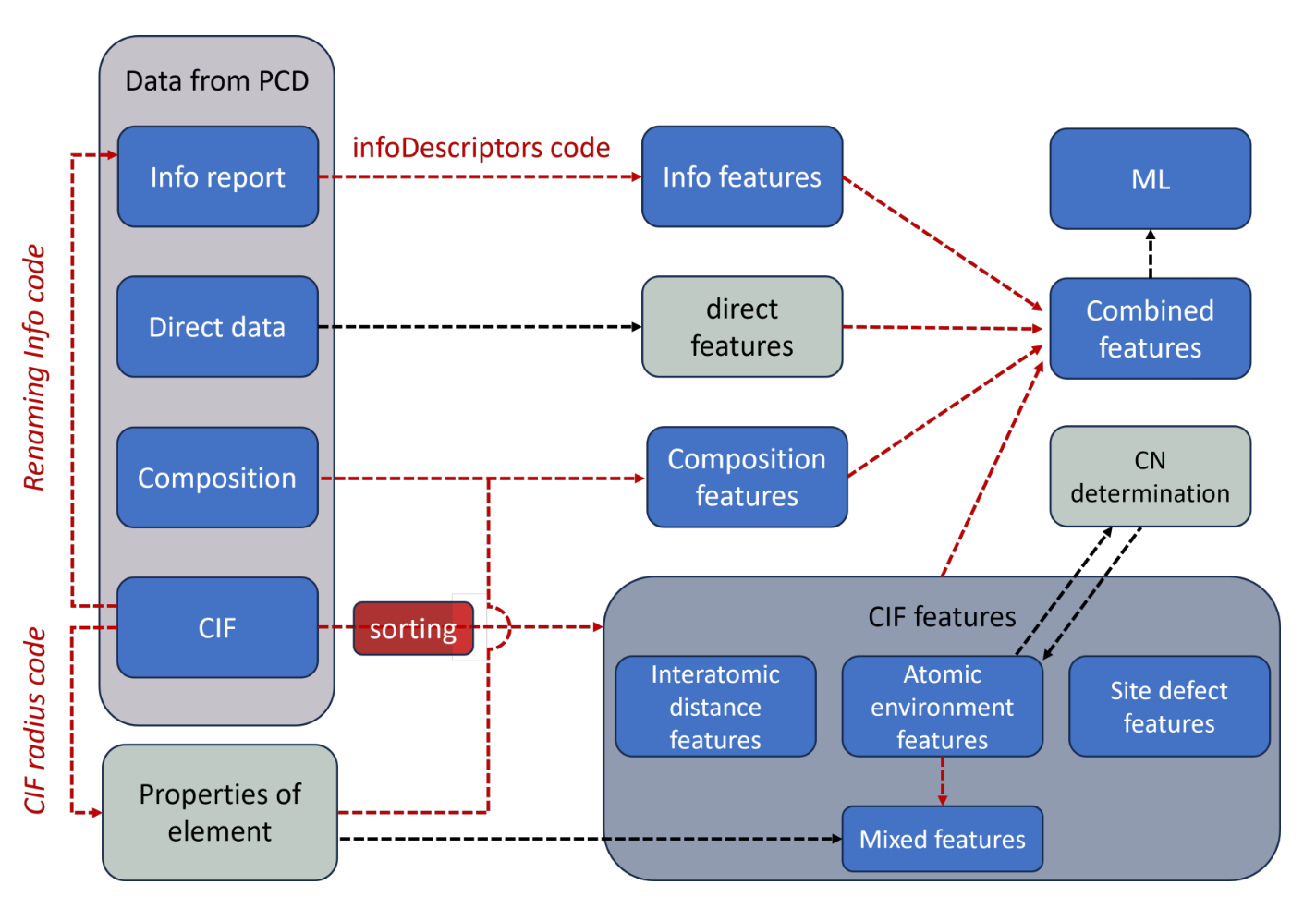
In Summer 2023, in collaboration with Dr. Anton Oliynyk, we developed an open-source Python tool crafted for the high-throughput extraction of crystal structure features. Traditional ML models in solid-state materials have predominantly relied on composition data to predict properties. The lack of structural information in the model often leads to incomplete property mapping and predictive noise
This geometric featurizer systematically processes raw Crystallographic Information Files (CIF) to construct supercells and meticulously extracts a comprehensive suite of descriptors, such as coordination numbers, interatomic distances, and atomic environments. These descriptors are not mere extrapolations from generic values but are tailored to the material’s specific structure, reducing noise and enhancing the relevance of ML applications for binary and ternary compounds. The package has demonstrated its robustness, having processed more than 10,000 binary and ternary CIF files. Leveraging structure-property correlations, we believe this crystal structure featurizer can be a promising tool for increasing predictive capabilities for material property optimization.
GitHub | Journal of Alloys and Compounds
Keywords: CIF, geometric descriptors, crystal structure, feature engineering, solid-state
2. Machine learning descriptors in materials chemistry used in multiple experimentally validated studies: Oliynyk elemental property dataset
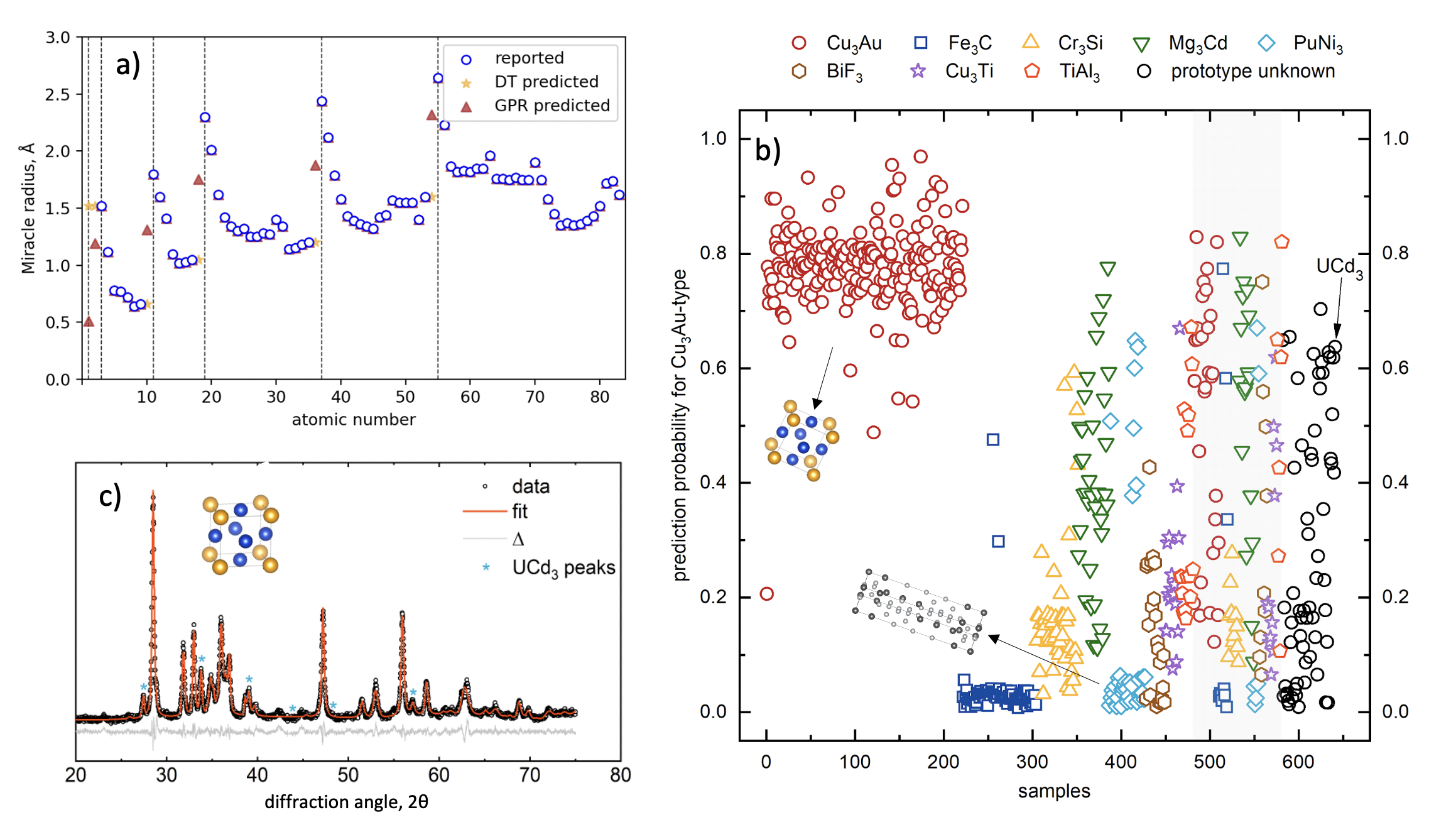
Materials informatics employs data-driven approaches for analysis and discovery of materials. Features also referred to as descriptors are essential in generating reliable and accurate machine-learning models. While general data can be obtained through public and commercial sources, features must be tailored to specific applications.
Common featurizers suitable for generic chemical problems may not be effective in features-property mapping in solid-state materials with ML models. Here, we have assembled the Oliynyk property list for compositional feature generation, which performs well on limited datasets (50 to 1,000 training data points) in the solid-state materials domain. The dataset contains 98 elemental features for atomic numbers from 1 to 92, including thermodynamic properties, electronic structure data, size, electronegativity, and bulk properties such as melting point, density, and conductivity. The dataset has been utilized peer-reviewed publications in predicting material hardness, classification, discovery of novel Heusler compounds, band gap prediction, and determining the site preference of atoms using machine learning models including support vector machines, random forests for classification, and support vector regression for regression problems. We have compiled the dataset by parsing data from publicly available databases and literature and further supplementing it by interpolating values with Gaussian process regression (GPR).
Data in Brief | Download dataset
Keywords: materials infomatics, feature engineering, GPR, elemental database, ML